Abstract
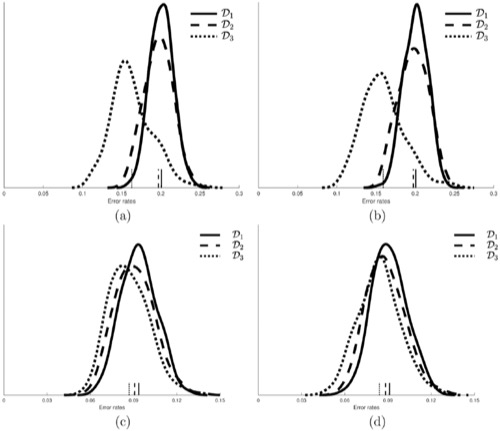
Features for standard expression microarray and RNA-Seq classification are expression averages over collections of cells. Single cell provides expression measurements for individual cells in a collection of cells from a particular tissue sample. Hence, it can yield feature vectors consisting of higher order and mixed moments. This article demonstrates the advantage of using these expression moments in cancer-related classification. We use synthetic data generated from 2 real networks, the mammalian cell cycle network and a melanoma-related pathway network, and real single-cell data generated via fluorescent protein reporters from 2 cell lines, HT-29 and HCT-116. The networks consist of hidden binary regulatory networks with Gaussian observations. The steady-state distributions of both the original and mutated networks are found, and data are drawn from these for moment-based classification using the mean, variance, skewness, and mixed moments. For the real data, we only observe 1 gene at a time, so that only the mean, variance, and skewness are considered, the analysis being done for 2 genes, EGFR and ERRB2. For the synthetic data, classification improves as we move from just the mean to mean, variance, and skewness and then to these plus the mixed moments. Comparisons are done with 3, 4, or 5 features, using feature selection. Sample size effects are considered. For the real data, we only consider mean, variance, and skewness, with results improving when the higher order moments are used as features.